Aritmogena kardiomiopatija
| Aritmogena kardiomiopatija | |
|---|---|
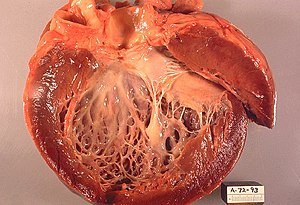 | |
| Presek kroz levu srčanu komoru, sa znacima subendokardialne fibroze | |
| Klasifikacija i spoljašnji resursi | |
| Specijalnost | кардиологија |
| Patient UK | Aritmogena kardiomiopatija |
| MeSH | D009202 |
[uredi na Vikipodacima] | |
Aritmogena kardiomiopatija (displazija AKM) primarno je oboljenje miokarda pri čemu je normalno tkivo miokarda zamenjeno fibromasnim, tokom procesa masne transformacije. Obično zahvata desnu srčanu komoru i mnogo je opasnija, jer može uzrokovati fatalne srčane aritmije tokom vežbanja. Ovo nasledno oboljenje čija prevalencija iznosi 1/1.000-1/5.000, klinički se ispoljava neishemijskom ventrikularnom aritmijom poreklom iz desne komore (DK).[1]
Simptomi bolesti su različiti, ali se najčešće manifestuju kao palpitacije, presinkopa i sinkopa, a neretko je iznenadna srčana smrt je prvi znak bolesti, uglavnom kod mladih ljudi i sportista.[1]
Postavljanje dijagnoze aritmogene kardiomiopatije (displazije) može biti veoma teško usled elektrokardiografskih specifičnosti, različite etiologije potencijalnih ventrikularnih aritmija s morfologijom bloka leve grane, procene funkcije i strukture desne komore, kao i tumačenja nalaza endomiokardnih biopsija. Za postavljanje dijagnoze aritmogene kardiomiopatije desne komore koriste se brojni klinički testovi: elektrokardiogram, ehokardiografija, perfuziona scintigrafija srca, nuklearna magnetna rezonancija, ventrikulografija, biopsija miokarda i genetski testovi.[2]
U terapiji aritmogene kardiomiopatije (displazije) koriste se beta blokatori, antiaritmijski lekovi, kateter ablacija i implantabilni kardioverter defibrilator, kao najefikasniji u sprečavanju iznenadne srčane smrti.[1]
Skrining pre aktivnog bavljenja sportom pokazao se kao efikasan u otkrivanju asimptomatskih pacijenata, čime je opao broj iznenadnih smrti kod mladih sportista.[1]
Istorija
Aritmogenu kardiomiopatije je prvi put opisao Giovani Maria Lancisi 1736. godine u svojoj knjizi De Motu Cordis et Aneurysmatibus kao porodično oboljenje koje se ponovilo u četiri generacije, a manifestovalo se palpitacijama, srčanom slabošću, dilatacijom i aneurizmom desne komore s iznenadnom smrću.[1][3]
Nakon opisa Giovani Maria Lancisi bilo je potrebno više od dva veka da bi Markus i saradnici 1982. godine opisali patološki supstrat tog oboljenja.[4]
Autozomno dominantni obrazac nasleđivanja promenljive penetracije prvi su opisali Nava i saradnici 1987. godine,[5] a prvi genski lokus (ARVD 1) otkrili su Rampazzo i saradnici 1994. godine na hromozomu 14q23.[6]
Svetska zdravstvena organizacija je aritmogenu kardiomiopatiju dene komore ( akronim ARVC/D) svrstala u kategoriju kardiomiopatija 1995. godine.[7]
Epidemiologija
- Morbiditet/mortalitet
Kako se mnogi slučajevi aritmogene kardiomiopatije otkriju tek tokom obdukcije njenu tačnu prevalenciju je teško utvrditi. Na osnovu većina izveštajakoji potiču iz Sjedinjenih Američkih Država i Evrope njena procenjena prevalneca iznosi 1/1.000 – 1/5.000.[8]
- Polne razlike
Oboleli od aritmogene kardiomiopatije pretežno su muškarci, sa muško : ženskim odnosom 2,7 : 1.[9]
- Starost
Prosečno doba nastanka prvih simptoma aritmogene kardiomiopatije su kasne dvadesete i rane tridesete godine života. Bolest obično nastaje posle puberteta, mada je bolest opisana i kod osoba svih uzrasta.
Etiologija
Uzrok aritmogene kardiomiopatije je nepoznat. Javlja se kod otprilike 1 od 5.000 ljudi. Iako se javlja kod onih bez porodične istorije, češča je sa porodičnom istorijom. Porodična istorija aritmogene kardiomiopatije prisutna je u najmanje 30 do 50%slučajeva. Zbog toga se preporučuje da se svi članovi porodice prvog i drugog stepena (roditelji, braća i sestre, deca, unuci, ujak, tetka, nećak, nećaka) pažljivo procene na ovaj oblik kardiomiopatije, čak i u odsustvu simptoma.[10]
Patofiziologija
Patofiziologiju aritmogene kardiomiopatije desne komore karakteriše progresivna zamena normalnog tkiva srčanog mišiča (miokarda) desne komore fibromasnim tkivom. Prednji infundibulum, vrh i dijafragmalni zid desne komore predilekciona su mesta transformacije tkiva, a poznata su pod nazivom „trougao displazije”.[11] Ova displazija može prouzrokovati dilataciju ili aneurizmu desne komore s pratećim paradoksnim pokretima. Za razliku od desne komore, leva komora i septum obično su pošteđeni fibromasne transformacije, ali mogu biti obuhvaćeni u slučajevima obimnog zahvatanja.
Promene na sprovodnom sistemu srca ometaju unutarkomorsko provođenje električnog impulsa što dovodi do:
- kasnih potencijala,
- epsilon talasa,
- bloka desne grane,
- nastanka fenomena reentri koji izaziva ventrikularne aritmije.[12]
Karakterističan elektrokardiografski nalaz (EKG) i aritmije nastaju usled disperzije miocita koji podstiču tahikardnu aktivnost kako displazija napreduje.[1]
Obliici fibromasne zamene
Postoje dva oblika fibromasne zamene:[1]
- Fibrolipomatozi tip 1, koji se opisuje i kao tipična bolest aritmogene kardiomiopatije u kome je predominantan nalaz masna zamena normalnog tkiva malom količinom fibroznog tkiva koje okružuje preostale „preživele” miokardne ćelije.
- Fibrolipomatozi tip 2, koja više odgovara opisu kardiomiopatije. Kod ovog tipa znatno je veća količina fibroznog tkiva nego masnog.[13]
Genetika
Aritmogene kardiomiopatije je genetsko oboljenje u oko 50% slučajeva.[10] Osnovni model nasleđivanja je autozomno dominantni s nepotpunom penetracijom,[14] mada postoji i autozomno recesivni tip nasleđivanja koji se manifestuje kao Naxos oboljenje, a koji pored aritmogene kardiomiopatije karakteriše palmoplantarna keratoza i ućebana kosa.[15] Upravo je taj recesivni model nasleđivanja pomogao u mapiranju gena odgovornih za aritmogene kardiomiopatije. Nakon što je istraživanjima ustanovljeno da epidermalne ćelije i miociti imaju sličan mehanički junkcioni aparat kao što su, recimo, dezmozomi i fascia adherens, došlo se do otkrića da gen koji kodira protein koji učestvuje u međućelijskom povezivanju odgovoran za nastanak aritmogene kardiomiopatije.[16][17]
| Vrsta | OMIM | Gen | Lokacija | Izvori |
|---|---|---|---|---|
| ARVD1lpl | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 107970 | TGFB3 | 14q23-q24 | [18] |
| ARVD2 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 600996 | RYR2 | 1q42-q43 | [19] |
| ARVD3 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 602086 | ? | 14q12-q22 | [20] |
| ARVD4 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 602087 | ? | 2q32.1-q32.3 | [21] |
| ARVD5 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 604400 | TMEM43 | 3p23 | [22][23] |
| ARVD6 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 604401 | ? | 10p14-p12 | [24] |
| ARVD7 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 609160 | DES | 10q22.3 | [25][26] |
| ARVD8 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 607450 | DSP | 6p24 | [27] |
| ARVD9 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 609040 | PKP2 | 12p11 | [28] |
| ARVD10 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 610193 | DSG2 | 18q12.1-q12 | [29][30][31] |
| ARVD11 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 610476 | DSC2 | 18q12.1 | [32][33][34] |
| ARVD12 | Online Mendelovsko nasleđivanje kod čoveka (OMIM) 611528 | JUP | 17q21 | [35][36] |
| ILK | 11p15.4 | [37] | ||
| LMNA | [38] |
Napredovanjem molekularne genetike, utvrđeno je da je aritmogene kardiomiopatije dezmozomalna bolest koja nastaje nakon defekata proteina ćelijske adhezije kao što su plakoglobin, dezmoplakin, dezmokolin, plakofilin-2, dezmoplein-2, transmembranski protein 43.[39]
Aritmogena kardiomiopatija se povezuje i s drugim genima koji nisu deo kompleksa ćelijske adhezije, npr. oni koji kodiraju srčani rijanodin receptor (RYR2) koji je odgovoran za oslobađanje kalcijuma iz sarkoplazmatičnog retikuluma, kao i geni za transformišući faktor rasta (TGFB3) što reguliše produkciju komponenti ekstracelularnog matriksa i menja ekspresiju gena koji kodiraju dezmozomalne proteine.[40]
Četiri dodatna gena povezana s autozomnom dominantnom aritmogenom kardiomiopatijom mapirana su, ali nisu identifikovana.[41]
Klinička slika
Klinička manifestacija aritmogene kardiomiopatije zavise od toga da li je simptomatska ili asimptomatska. Ukoliko je aritmogene kardiomiopatija asimptomatska, bolest teško se prepoznaje, osim u slučajevima kada lekar poseduje podatak o njenoj porodičnoj istoriji. Iako gotovo svi skrining programi sportista obuhvataju rutinski fizikalni pregled i istoriju bolesti, čak i u slučajevima koji obuhvataju EKG, lekar mora biti obazriv u pogledu nespecifičnih EKG promena koje se susreću kod aritmogene kardiomiopatije.[42] Tipičan primer obolele osobe sa aritmogenom kardiomiopatije jeste mladi čovek koji se žali na umor, tahikardiju, palpitacije, ponekad bol u grudima, presinkopu i sinkopu.
Vrlo često je iznenadna srčana smrt prva manifestacija aritmogene kardiomiopatije kod sportista, ali i radnika koji se bave napornim fizičkim radom.[43] To je najbolje dokumentovano u Italiji, gde je aritmogene kardiomiopatije bila uzrok u 20% slučajeva iznenadne srčane smrti ljudi mlađih od 35 godina.[44]
Prevencija iznenadne srčane smrti
Kako je srčani zastoj kod aritmogene kardiomiopatije posledica udruženog delovnja različitih faktora (osnove same bolesti, provocirajućih faktora, aritmija) preventivne mere su usmere upravo na njih.
Svaki napor koji izazove opterećenje i istezanje miokarda desne komore smatra se potencijalnim okidačem za ishemijsku bolest srca. Sportska aktivnost pet puta povećava rizik od ishemijske bolesti srca kod mladih.[44] Stoga je kod osoba s asimptomatskom aritmogenom kardiomiopatijom od krucijalnog je značaja izbegavanje napora. U tom smislu kada se jednom postavi dijagnoza aritmogene kardiomiopatije, bolesnik se ne sme više baviti takmičarskim sportovima ili drugim intenzivnim, napornim fizičkim aktivnostima.[45][46]
- Skrining pre aktivnog bavljenja sportom
Skrining pre aktivnog bavljenja sportom, koji je sa sobom doneo sportske diskvalifikacije, pokazao se kao veoma efikasan u sprečavanju iznenadne srčane smrti. Na osnovu iskustva iz Italije, pre uvođenja obaveznog skrininga, stopa iznenadne srčane smrti bila je 1/28.000, a nakon uvođenja 1/250.000 godišnje, uglavnom zbog identifikacije i diskvalifikacije pacijenata sa aritmogenom kardiomiopatijom.[47]
Najbolji način da se spreči iznenadna srčana smrt od ARVC svakako je radikalna forma koja se može postići na sledeće načine:[1]
- Transplantacijom srca u slučajevima refraktarne srčane slabosti i/ili aritmija.
- Savremenom (zasad nepoznatom) terapijom koja bi sprečila apoptozu miocita na molekularnom nivou.[48]
Dijagnoza
Za postavljanje dijagnoze aritmogene kardiomiopatije koriste se sledeći testovi: elektrokardiogram, ehokardiografija, perfuziona scintigrafija srca, nuklearna magnetna rezonancija, ventrikulografija, biopsija miokarda i moždani natrijuretski peptid.
- Elektrokardiogram

EKG u mirovanju u 50–90% slučajeva sa aritmogenom kardiomiopatijom, pokazaće sledeće tipične promene:[1]
- Inverzija T-talasa V1–V4;
- Kašnjenja provođenja zbog inkompletnog ili kompletnog bloka desne grane;
- Ventrikularne ekstrasistole s blokom leve grane i negativnim ili neodređenim kompleksom QRS u II, III i aVF odvodu i pozitivnim u aVL odvodu;
- Ventrikularni postekscitacioni talasi – epsilon talas.
- Ehokardiografija
Često je prvi test taj koji pokazuje karakteristične abnormalnosti za aritmogenu kardiomiopatiju iako su podaci o njegovoj preciznosti varijabilni.[49] Normalni ehokardiografski pregled ne isključuje aritmogenu kardiomiopatiju, s obzirom na to da rane forme bolesti ne moraju biti odmah uočljive. S druge strane, ehokardiografski verifikovana regionalna ili difuzno uvećana desna komora ili njena disfunkcija kod bolesnika sa sumnjom na aritmogenu kardiomiopatiju jeste čvrst dokaz oboljenja. Međutim, pre postavljanja dijagnoze aritmogene kardiomiopatije, neophodno je isključiti druge uzroke uvećanja desne komore, kao što su šantovi, kongenitalne ili valvularne bolesti.[50]
- Perfuziona scintigrafija srca
Predstavlja alternativu ventrikulografiji i ehokardiografiji u slučajevima gde je prednju lokalizaciju i iregularnost oblika desne komore teško proceniti. Pojedini istraživači koriste tu metodu na testu opterećenja kako bi procenili kinetiku zida desne komore, a njena disfunkcija čvrsto upućuje na dijagnozu aritmogene kardiomiopatije.[51]
- Nuklearna magnetna rezonancija

Nuklearna magnetna rezonancija (NMR) smatra se zlatnim standardom za neinvazivno postavljanje dijagnoze aritmogene kardiomiopatije. NMR može detektovati masnu infiltraciju ili istanjenje infundibuluma, kao i dijafragmalni zid desne komore, koji se teško vizualizuje ehokardiografskim pregledom.[52] Takođe, NMR se može koristiti za korelaciju morfoloških nalaza tankog zida ili aortne aneurizme s diskinetičkim segmentima desne komore putem gradijent eho pulsne sekvence.[53] Pojedini centri kao alternativu NMR koriste specifičnu formu kompjuterizovane tomografije s elektronskim snopom, čime se postiže podjednaka vizualizacija promena kao i magnetnom rezonancijom.[54]
Moždani natrijuretski peptid
Lučenje moždanog natrijuretskog peptida (BNP) kao odgovora na aritmogenu kardiomiopatiju predstavlja koristan dijagnostički i prognostički marker. Merenjem nivo tog peptida u kliničkim studijama u cilju razlikovanja aritmogene kardiomiopatije od drugih formi idiopatskih ventrikularnih tahikardija,[55] utvrđeno je da je nivo tog peptida u obrnutom odnosu s ejekcionom frakcijom desne srčane komore, što se može koristiti kao potencijalni metod praćenja progresije aritmogene kardiomiopatije desne komore.[1]
Angiografija desne komore
Pre rutinskog uvođenja ehokardiografije kao standardne procedure za postavljanje dijagnoze aritmogene kardiomiopatije, ventrikulografija je često korišćena kao pouzdan test za lokalizaciju abnormalnosti kao što su proširenja, dilatacija i disfunkcija desne komore.
Danas se, ehokardiografija pokazala kao ekvivalentna metoda ventrikulografiji u pogledu kvaliteta slika, ali i kao manje invazivna, jeftinija, dostupnija i lakša za izvođenje, što je angiografija desne komore, kao dijagnostičku metodu skoro u potpunosti potisnulo.[1]
Biopsija miokarda

Zlatni standard u postavljanju dijagnoze aritmogene kardiomiopatije svakako je transmuralna biopsija uzoraka prilikom obdukcije ili transplantacije srca. Ubraja se u manje senzitivne dijagnostičke testove za aritmogene kardiomiopatije, ali se može primeniti kod svih pacijenata sa sumnjom na aritmogenu kardiomiopatiju.[1]
Na osnovu predloženih histomorfometrijskih kriterijuma za evaluaciju uzoraka biopsije, za postavljanje dijagnoze aritmogene kardiomiopatije neophodno je da bude manje od 45% rezidualnih miocita, više od 40% fibroznog i tri posto masnog tkiva. Senzitivnost tih kriterijuma je 67%, a specifičnost 92%.[1]
Terapija
Lečenje aritmogene kardiomiopatije je kontroverzno jer obuhvata više opcija: antiaritmike, radiofrekventnu ablaciju, implantabilni kardioverter defibrilator, hirurške metode, transplantaciju srca.[1]
Antiaritmici
Najveća studija sprovedena na 81 bolesniku otkrila je da je sotalol najefikasniji lek u lečenju aritmogene kardiomiopatije s uspešnošću od 68%, za razliku od amiodarona nakon koga je je uspešnost lečenja bila 26%.[56] Zaključeno je da ukoliko je terapija sotalolom neuspešna, neophodno je razmišljati o invazivnijem načinu lečenja.[1]
Radiofrekventna kateter ablacija
Radiofrekventna kateter ablacija efikasna je za neke pacijente s ventrikularnom tahikardijom refraktarnom na antiaritmike.[1] Iako takav način lečenja može biti efektivan u kratkom periodu, sama procedura povezana je s visokom stopom ponovnog javlja- nja (kod 40% slučajeva ponovi se za tri godine), što ukazuje na to da je taj način zbrinjavanja aritmogene kardiomiopatije palijativne prirode.[57]
Implantabilni kardioverter defibrilator
Implantabilni kardioverter defibrilator (akronim ICD) – Uloga ICD je da konvertuje ventrikularni flater/fibrilaciju u sinusni ritam. Corrado i saradnici su tokom 48-mesečnog praćenja pacijenata s ugrađenim ICD dokazali da je 76% pacijenata bilo oslobođeno električnog šoka u slučajevima ventrikularnog flatera/fibrilacije, sa stopom preživljavanja 96% u istom periodu.[58]
Ukoliko bismo uzeli u razmatranje da bi svaka epizoda električnog šoka bila praćena smrtnim ishodom, ICD je spasao 20% pacijenata.[1][59]
Hirurške metode
Hirurški pristup lečenju aritmogene kardiomiopatije indikuje odvajanje (disartikulaciju) desne i leve komore, čime se te komore izoluju, što sprečava širenje ventrikularne tahikardije nastale u desnoj komori na levu komoru.[60] Taj metod praćen je brzom akutnom dekompenzacijom desne komore, koja se posle progresivno oporavlja. Međutim, taj metod je praktično napušten uvođenjem kardioverter defibrilatora.[61]
Transplantacija srca
Indikacije za transplantaciju desne komore još nisu definisane, za razliku od levostrane kardiomiopatije, gde su kriterijumi jasni.[62] S obzirom na to da je desnostrana kardiomiopatija redak nalaz, nepotpuno je znanje o desnom delu srca i plućnoj hemodinamici, naročito tokom aritmogene aktivnosti.[1][63]
Zasad je transplantacija srca krajnji način lečenja aritmogene kardiomiopatije u slučajevima kad postoji obostrana progresivna komorska slabost s porastom broja ventrikularnih tahikardija.[1][64]
Prognoza
Prirodni tok aritmogene kardiomiopatije prolazi kroz četiri faze:[1]
| Faze | Karakteristike |
|---|---|
| Prva „skrivena” faza | Obično je bez simptoma. U ovoj fazi pacijenti su u velikom riziku od iznenadne srčane smrti, naročito tokom vežbanja. Ukoliko postoje strukturne promene, one su malog obima i obično se nalaze u „trouglu” displazije.[1] |
| Druga, „otvorena” faza | Karakteriše se simptomatskom ventrikularnom aritmijom, i uočljivim morfološkim i funkcionalne promene desne komore.[1] |
| Treća faza | Odlikuje se difuznim oštećenjem desne komore, dok je leva komora očuvana.[1] |
| Četvrta, „napredna” faza | U ovoj fazi nastaje ozbiljno, difuzno biventrikularno obuhvatanje, s fenotipskom ekspresijom koja podseća na dilatativnu kardiomiopatiju.[1][65][8] |
Izvori
- ^ а б в г д ђ е ж з и ј к л љ м н њ о п р с т ћ у ф Sanja MAZIĆ , Biljana LAZOVIĆ i Marina ĐELIĆ, Aritmogena kardiomiopatija desne komore kao uzrok iznenadne srčane smrti mladih - pregled literature. Med Pregl 2012; LXV (9-10): 396-404. Novi Sad: septembar-oktobar
- ^ „Arrhythmogenic Cardiomyopathy | Cardiomyopathy UK”. www.cardiomyopathy.org. Приступљено 2022-04-13.
- ^ Norman, M. W.; McKenna, W. J. (1999). „Arrhythmogenic right ventricular cardiomyopathy/dysplasia: perspectives on diseases”. Z Kardiol. 88: 550—4.
- ^ Marcus F, Fontaine G, Guirdaudon G, Frank R, Laurenceau JL, Malergue C; et al. (1982). „Right ventricular dysplasia: a report of 24 adult cases”. Circulation. 65: 384—98. CS1 одржавање: Вишеструка имена: списак аутора (веза)
- ^ Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja GF; et al. (1988). „Familial occurrence of right ventricular dysplasia: a study involving nine families”. Journal of the American College of Cardiology. 12: 1222—8. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G; et al. (1994). „The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24”. Hum Mol Genet. 3: 959—62. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ „Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies.”. Heart. 44 (6): 672—673. 1980-12-01. ISSN 1355-6037. doi:10.1136/hrt.44.6.672.
- ^ а б Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J; et al. (2005). „Arrhythmogenic right ventricular dysplasia: a United States experience”. Circulation. 112: 3823—32. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Szymański P, Klisiewicz A, Hoffman P (2011). „ARVC/D task force imaging criteria: it is difficult to get along with the guidelines”. JACC Cardiovasc Imaging. 4 (6): 686. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ а б „Arrhythmogenic Right Ventricular Dysplasia (ARVD)”. Cleveland Clinic. Приступљено 2022-04-14.
- ^ O’Donnell D, Cox D, Bourke J, MitchellL, Furniss S (2003). „Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract”. European Heart Journal. 24: 801—10. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy: just a matter of fat? Cardiovasc Pathol. 14: 37—41. 2005. Недостаје или је празан параметар
|title=(помоћ). - ^ Tandri, H.; Saranathan, M.; Rodriguez, E. R.; et al. (2005). „Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging.”. Journal of the American College of Cardiology. 45: 98—103.
- ^ Sen-Chowdhry S, Syrris P, McKenna WJ (2005). „Genetics of right ventricular cardiomyopathy.”. Journal of Cardiovascular Electrophysiology. 16: 927—35. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Protonotarios, N.; Tsatsopoulou, A. (2004). „Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy”. Cardiovasc Pathol. 13: 185—94.
- ^ McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A; et al. (2000). „Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease).”. Lancet. 355: 2119—24. CS1 одржавање: Вишеструка имена: списак аутора (веза)
- ^ Rampazzo, A.; Nava, A.; Malacrida, S.; et al. (2002). „Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy.”. Am J Hum Genet. 71: 1200—6. .
- ^ Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A (фебруар 2005). „Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1”. Cardiovascular Research. 65 (2): 366—373. PMID 15639475. doi:10.1016/j.cardiores.2004.10.005
 . CS1 одржавање: Формат датума (веза)
. CS1 одржавање: Формат датума (веза) - ^ Milting H, Lukas N, Klauke B, Körfer R, Perrot A, Osterziel KJ, Vogt J, Peters S, Thieleczek R, Varsányi M (август 2006). „Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy”. Cardiovascular Research. 71 (3): 496—505. PMID 16769042. doi:10.1016/j.cardiores.2006.04.004 . CS1 одржавање: Формат датума (веза)
- ^ Severini GM, Krajinovic M, Pinamonti B, Sinagra G, Fioretti P, Brunazzi MC, Falaschi A, Camerini F, Giacca M, Mestroni L (јануар 1996). „A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14”. Genomics. 31 (2): 193—200. PMID 8824801. doi:10.1006/geno.1996.0031. hdl:1765/58364. CS1 одржавање: Формат датума (веза)
- ^ Rampazzo A, Nava A, Miorin M, Fonderico P, Pope B, Tiso N, Livolsi B, Zimbello R, Thiene G, Danieli GA (октобар 1997). „ARVD4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chromosome 2 long arm”. Genomics. 45 (2): 259—263. PMID 9344647. doi:10.1006/geno.1997.4927. hdl:11577/2461314. CS1 одржавање: Формат датума (веза)
- ^ Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL (април 2008). „Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene”. American Journal of Human Genetics. 82 (4): 809—821. PMC 2427209 . PMID 18313022. doi:10.1016/j.ajhg.2008.01.010. CS1 одржавање: Формат датума (веза)
- ^ Christensen AH, Andersen CB, Tybjaerg-Hansen A, Haunso S, Svendsen JH (септембар 2011). „Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy”. Clinical Genetics. 80 (3): 256—264. PMID 21214875. S2CID 5617616. doi:10.1111/j.1399-0004.2011.01623.x. CS1 одржавање: Формат датума (веза)
- ^ Li D, Ahmad F, Gardner MJ, Weilbaecher D, Hill R, Karibe A, Gonzalez O, Tapscott T, Sharratt GP, Bachinski LL, Roberts R (јануар 2000). „The locus of a novel gene responsible for arrhythmogenic right-ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14”. American Journal of Human Genetics. 66 (1): 148—156. PMC 1288320 . PMID 10631146. doi:10.1086/302713. CS1 одржавање: Формат датума (веза)
- ^ Protonotarios A, Brodehl A, Asimaki A, Jager J, Quinn E, Stanasiuk C, Ratnavadivel S, Futema M, Akhtar MM, Gossios TD, Ashworth M, Savvatis K, Walhorn V, Anselmetti D, Elliott PM, Syrris P, Milting H, Lopes LR (јун 2021). „The Novel Desmin Variant p.Leu115Ile Is Associated With a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy”. The Canadian Journal of Cardiology. 37 (6): 857—866. PMID 33290826. S2CID 228078648. doi:10.1016/j.cjca.2020.11.017. CS1 одржавање: Формат датума (веза)
- ^ Bermúdez-Jiménez FJ, Carriel V, Brodehl A, Alaminos M, Campos A, Schirmer I, Milting H, Abril BÁ, Álvarez M, López-Fernández S, García-Giustiniani D, Monserrat L, Tercedor L, Jiménez-Jáimez J (април 2018). „Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia”. Circulation. 137 (15): 1595—1610. PMID 29212896. doi:10.1161/CIRCULATIONAHA.117.028719 . hdl:10481/89514 . CS1 одржавање: Формат датума (веза)
- ^ Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A (август 2005). „Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations”. European Heart Journal. 26 (16): 1666—1675. PMID 15941723. doi:10.1093/eurheartj/ehi341 . CS1 одржавање: Формат датума (веза)
- ^ Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Drenckhahn J, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L (новембар 2004). „Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy”. Nature Genetics. 36 (11): 1162—1164. PMID 15489853. doi:10.1038/ng1461 . CS1 одржавање: Формат датума (веза)
- ^ Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A (март 2006). „Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy”. Circulation. 113 (9): 1171—1179. PMID 16505173. doi:10.1161/CIRCULATIONAHA.105.583674 . CS1 одржавање: Формат датума (веза)
- ^ Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russell SD, Bluemke DA, Dietz HC, Calkins H, Judge DP (јул 2006). „DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy”. American Journal of Human Genetics. 79 (1): 136—142. PMC 1474134 . PMID 16773573. doi:10.1086/504393. CS1 одржавање: Формат датума (веза)
- ^ Brodehl A, Meshkov A, Myasnikov R, Kiseleva A, Kulikova O, Klauke B, Sotnikova E, Stanasiuk C, Divashuk M, Pohl GM, Kudryavtseva M, Klingel K, Gerull B, Zharikova A, Gummert J, Koretskiy S, Schubert S, Mershina E, Gärtner A, Pilus P, Laser KT, Sinitsyn V, Boytsov S, Drapkina O, Milting H (април 2021). „Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset”. International Journal of Molecular Sciences. 22 (7): 3786. PMC 8038858 . PMID 33917638. doi:10.3390/ijms22073786 . CS1 одржавање: Формат датума (веза)
- ^ Brodehl A, Weiss J, Debus JD, Stanasiuk C, Klauke B, Deutsch MA, Fox H, Bax J, Ebbinghaus H, Gärtner A, Tiesmeier J, Laser T, Peterschröder A, Gerull B, Gummert J, Paluszkiewicz L, Milting H (април 2020). „A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy”. Journal of Molecular and Cellular Cardiology. 141: 17—29. PMID 32201174. doi:10.1016/j.yjmcc.2020.03.006 . CS1 одржавање: Формат датума (веза)
- ^ Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, MacRae CA, Gerull B (децембар 2006). „Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy”. American Journal of Human Genetics. 79 (6): 1081—1088. PMC 1698714 . PMID 17186466. doi:10.1086/509044. CS1 одржавање: Формат датума (веза)
- ^ Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ (новембар 2006). „Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2”. American Journal of Human Genetics. 79 (5): 978—984. PMC 1698574 . PMID 17033975. doi:10.1086/509122. CS1 одржавање: Формат датума (веза)
- ^ Antoniades L, Tsatsopoulou A, Anastasakis A, Syrris P, Asimaki A, Panagiotakos D, Zambartas C, Stefanadis C, McKenna WJ, Protonotarios N (септембар 2006). „Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin-2 and plakoglobin (Naxos disease) in families from Greece and Cyprus: genotype-phenotype relations, diagnostic features and prognosis”. European Heart Journal. 27 (18): 2208—2216. PMID 16893920. doi:10.1093/eurheartj/ehl184. CS1 одржавање: Формат датума (веза)
- ^ Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ (новембар 2007). „A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy”. American Journal of Human Genetics. 81 (5): 964—973. PMC 2265660 . PMID 17924338. doi:10.1086/521633. CS1 одржавање: Формат датума (веза)
- ^ Brodehl A, Rezazadeh S, Williams T, Munsie NM, Liedtke D, Oh T, Ferrier R, Shen Y, Jones SJ, Stiegler AL, Boggon TJ, Duff HJ, Friedman JM, Gibson WT, Childs SJ, Gerull B (јун 2019). „Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy”. Translational Research. 208: 15—29. PMC 7412573 . PMID 30802431. doi:10.1016/j.trsl.2019.02.004. CS1 одржавање: Формат датума (веза)
- ^ Forleo C, Carmosino M, Resta N, Rampazzo A, Valecce R, Sorrentino S, Iacoviello M, Pisani F, Procino G, Gerbino A, Scardapane A, Simone C, Calore M, Torretta S, Svelto M, Favale S (2015). „Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy”. PLOS ONE. 10 (4): e0121723. Bibcode:2015PLoSO..1021723F. PMC 4383583 . PMID 25837155. doi:10.1371/journal.pone.0121723 .
- ^ Corrado, D.; Thiene, G. (2006). „Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies.”. Circulation. 113: 1634—7. .
- ^ Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C; et al. (2005). „Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1”. Cardiovascular Research. 65: 366—73. CS1 одржавање: Вишеструка имена: списак аутора (веза)
- ^ Burkett, E.; Hershberger, R. E. (2005). „State of the art: clinical and genetic issues in familial dilated cardiomyopathy”. Journal of the American College of Cardiology. 45: 969—81. .
- ^ Calkins, H. (2006). „Arrhythmogenic right-ventricular dysplasia/cardiomyopathy.”. Curr Opin Cardiol. 21: 55—63.
- ^ Aranđelović A, Pavlović S, Mazić S, Aleksandrić B (2004). „Naprasna srčana smrt sportista”. Srp Arh Celok Lek. 132: 194—7. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ а б Pelliccia A, Corrado D, Bjørnstad HH, Panhuyzen-Goedkoop N, Urhausen A, Carre F. „Recommendations for participation in competitive sport and leisure-time physical activity in individuals with cardiomyopathies, myocarditis and pericarditis.”. Eur J Cardiovasc Prev Rehabil. 13 (6): 876—85. 2006. .
- ^ Mezzani A, Agostoni P, Cohen-Solal A, Corrà U, Jegier A, Kouidi E. „Standards for the use of cardiopulmonary exercise testing for the functional evaluation of cardiac patients: a report from the Exercise Physiology Section of the European Association for Cardiovascular Prevention and Rehabilitation.”. Eur J Cardiovasc Prev Rehabil. 16 (3): 249—67. 2009. .
- ^ Deyell, M. W.; Andrade, J. G.; McManus, B. M.; Leipsic, J. (2011). „The other side of arrhythmogenic right ventricular cardiomyopathy.”. Can J Cardiol. 27 (2): 263. .e13-6
- ^ Gemayel C, Pelliccia A, Thompson PD. (2001). „Arrhythmogenic right ventricular cardiomyopathy.”. Journal of the American College of Cardiology. 38 (7): 1773—81. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S; et al. (2006). „Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy.”. Circ Res. 99: 646—55. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Marcus F, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA; et al. (2010). „Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria”. Circulation. 121 (13): 1533—41. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Gimeno JR, Lacunza J, García-Alberola A, Cerdán MC, Oliva MJ, García-Molina E. „Penetrance and risk profile in inherited cardiac diseases studied in a dedicated screening clinic”. American Journal of Cardiology. 104 (3): 406—10. 2009. .
- ^ Lindström L, Nylander E, Larsson H, Wranne B (2005). „Left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy: a scintigraphic and echocardiographic study”. Clin Physiol Funct Imaging. 25 (3): 171—7. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Vermes E, Strohm O, Otmani A, Childs H, Duff H, Friedrich MG (2011). „Impact of the revision of arrhythmogenic right ventricular cardiomyopathy/dysplasia task force criteria on its prevalence by CMR criteria”. JACC Cardiovasc Imaging. 4 (3): 282—7. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Santangeli P, Pieroni M, Pieroni M, Dello Russo A, Casella M, Pelargonio G; et al. (2010). „Noninvasive diagnosis of electroanatomic abnormalities in arrhythmogenic right ventricular cardiomyopathy”. Circ Arrhythm Electrophysiol. 3 (6): 632—8. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Kimura F, Sakai F, Sakomura Y, Fujimura M, Ueno E, Matsuda N; et al. (2002). „Helical CT features of arrhythmogenic rightm ventricular cardiomyopathy”. Radiographics. 22 (5): 1111—24. CS1 одржавање: Вишеструка имена: списак аутора (веза)
- ^ Matsuo K, Nishikimi T, Yutani C, Kurita T, Shimizu W, Taguchi A; et al. (1998). „Diagnostic value of plasma levels of brain natriuretic peptide in arrhythmogenic right ventricular dysplasia”. Circulation. 98: 2433—40. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Bomma, C.; Dalal, D.; Tandri, H.; et al. (2007). „Evolving role of multidetector computed tomography in evaluation of arrhythmogenic right ventricular dysplasia/cardiomyopathy”. American Journal of Cardiology. 100: 99—105. .
- ^ Polin, G. M.; Haqqani, H; Tzou, W.; Hutchinson, M. D.; Garcia, F. C.; Callans, D. J.; et al. (2011). „Endocardial unipolar voltage mapping to identify epicardial substrate in arrhythmogenic right ventricular cardiomyopathy/dysplasia.”. Heart Rhythm. 8 (1): 76—83. .
- ^ Komura, M.; et al. (2010). „Clinical course of arrhythmogenic right ventricular cardiomyopathy in the era of implantable cardioverter-defibrillators and radiofrequency catheter ablation.”. Int Heart J. 51 (1): 34—40. .
- ^ Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A; et al. (2003). „Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia.”. Circulation. 108: 3084—91. CS1 одржавање: Вишеструка имена: списак аутора (веза)
- ^ Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A; et al. (2003). „Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia.”. Circulation. 108: 3084—91. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Wichter, T.; Paul, T. M.; Eckardt, L.; et al. (2005). „Arrhythmogenic right ventricular cardiomyopathy: antiarrhythmi drugs, catheter ablation, or ICD?”. Herz. 30: 91—101.
- ^ Ikari, N. M.; Azeka, E; Aiello EVD; Atik, E; BarberoMarcial, M; Ebaid, M. (2001). „Uhl’s anomaly: differential diagnosis and indication for cardiac transplantation in an infant.”. Arq Bras Cardiol. 77: 69—76. .
- ^ Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G (2011). „Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry”. European Heart Journal. 32 (9): 1105—13. CS1 одржавање: Вишеструка имена: списак аутора (веза).
- ^ Gilljam, T.; Bergh, C. H. (2009). „Right ventricular cardiomyopathy: timing of heart transplantation in Uhl’s anomaly and arrythmogenic right ventricular cardiomyopathy”. European Heart Journal. 11: 106—9.
- ^ Corrado, D.; Basso, C.; Thiene, G. (2009). „Arrhythmogenic right ventricular cardiomyopathy: an update.”. Heart. 95: 766—73.

Spoljašnje veze
- 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy
 | Molimo Vas, obratite pažnju na važno upozorenje u vezi sa temama iz oblasti medicine (zdravlja). |
Aritmogena kardiomiopatija na srodnim projektima Vikipedije:







